| Basic Information | |
|---|---|
| Species | Brachypodium distachyon |
| Cazyme ID | Bradi2g58860.1 |
| Family | GT4 |
| Protein Properties | Length: 1079 Molecular Weight: 118893 Isoelectric Point: 6.6002 |
| Chromosome | Chromosome/Scaffold: 2 Start: 56638957 End: 56644118 |
| Description | sucrose phosphate synthase 3F |
| View CDS | |
| External Links |
|---|
| NCBI Taxonomy |
| Plaza |
| CAZyDB |
| Signature Domain Download full data set without filtering | |||
|---|---|---|---|
| Family | Start | End | Evalue |
| GT4 | 483 | 652 | 0 |
| TNPHKPMILALSRPDAKKNITTLVRAFGECRPLRELANLTLIMGNRDDIDEMPAGNANVLTTVLKLVDKYDLYGSVAFPKHHNQADVPEIYRLAAKMKGV FINPALVEPFGLTLIEAAAHGLPIVATKNGGPVDITTALNNGLLVDPHDKNAIADALLKLVADKNLWQEC | |||
| Full Sequence |
|---|
| Protein Sequence Length: 1079 Download |
| MAGNEWINGY LEAILDSGGA AAGGGAGGGD PKSAVAGAGA SSASPRGPHM NFSPTHYFVE 60 EVVKGVDESD LHRTWIKVVA TRNARERSTR LENMCWRIWH LARKKKQLEL EGIQRMSARQ 120 KEQEQVRREA TEDLAEDLSE GEKGDTVGEL ASYGTPKKKF QRNFSDLTVW SDDNKEKKLY 180 IVLISVHGLV RGENMELGSD SDTGGQVKYV VELARALSLM PGVYRVDLFT RQVSSPDVDW 240 SYGEPTEMLC SGSTDAEGGE SAGAYIVRIP CGPRDKYIKK EALWPYLQEF VDGALAHILN 300 MSRALGEQVG RGKPVLPYVI HGHYADAGDV ASLLSGALNV PMVLTGHSLG RNKLEQIMKQ 360 GRMSKEEIDS TYKIMRRIEG EELALDAAEL VITSTRQEID EQWGLYDGFD VKLEKVLRAR 420 TRRGVSCHGR FMPRMVVIPP GMDFSNVVAE DVDGDGDGKD DMLDGASPRS LPPIWAEVMR 480 FLTNPHKPMI LALSRPDAKK NITTLVRAFG ECRPLRELAN LTLIMGNRDD IDEMPAGNAN 540 VLTTVLKLVD KYDLYGSVAF PKHHNQADVP EIYRLAAKMK GVFINPALVE PFGLTLIEAA 600 AHGLPIVATK NGGPVDITTA LNNGLLVDPH DKNAIADALL KLVADKNLWQ ECRKNGLRNI 660 HLYSWPEHCR QYLTRVAGCR IRNPRWLTDT PADTGADEED ALEDSLIDFQ DLSLRLSIDG 720 ERGASLNEPA SSDPQDQVQK IMNKIKQSSS HAHPSGIPDG SGAGEGDVKS HSELASGGVN 780 KYPLLRRRRR LFIVAVDCYG DDGRATKKML QVIQEVFRAV RSDSQMSKIS GFALSTAMPL 840 SETLQLLQLG KVPPTDFDAL ICGSGSEVYY PGTAQCVDAQ GRLRPDQDYL LHINHRWSHD 900 GARQTIGKLM AHDGSSDAVE PDVESCNAHC VSFFVRDPKK VKTIDELRER LRMRGLRCHL 960 MYCRNSTRLQ VVPLMASRSQ ALRYLFVRWG LPVGNMFLIV GEHGDSDREE MLSGLHKTVI 1020 VQGVTEKGSE QLLRSSGSYH KEDVVPAVSP LTASTRGELK ADEIMRALKE VTKTSSGM* 1080 |
| Functional Domains Download unfiltered results here | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cdd ID | Domain | E-Value | Start | End | Length | Domain Description | ||
| cd03801 | GT1_YqgM_like | 3.0e-36 | 204 | 673 | 474 | + This family is most closely related to the GT1 family of glycosyltransferases and named after YqgM in Bacillus licheniformis about which little is known. Glycosyltransferases catalyze the transfer of sugar moieties from activated donor molecules to specific acceptor molecules, forming glycosidic bonds. The acceptor molecule can be a lipid, a protein, a heterocyclic compound, or another carbohydrate residue. This group of glycosyltransferases is most closely related to the previously defined glycosyltransferase family 1 (GT1). The members of this family may transfer UDP, ADP, GDP, or CMP linked sugars. The diverse enzymatic activities among members of this family reflect a wide range of biological functions. The protein structure available for this family has the GTB topology, one of the two protein topologies observed for nucleotide-sugar-dependent glycosyltransferases. GTB proteins have distinct N- and C- terminal domains each containing a typical Rossmann fold. The two domains have high structural homology despite minimal sequence homology. The large cleft that separates the two domains includes the catalytic center and permits a high degree of flexibility. The members of this family are found mainly in certain bacteria and archaea. | ||
| TIGR02470 | sucr_synth | 5.0e-47 | 181 | 674 | 538 | + sucrose synthase. This model represents sucrose synthase, an enzyme that, despite its name, generally uses rather produces sucrose. Sucrose plus UDP (or ADP) becomes D-fructose plus UDP-glucose (or ADP-glucose), which is then available for cell wall (or starch) biosynthesis. The enzyme is homologous to sucrose phosphate synthase, which catalyzes the penultimate step in sucrose synthesis. Sucrose synthase is found, so far, exclusively in plants and cyanobacteria [Energy metabolism, Biosynthesis and degradation of polysaccharides]. | ||
| cd03800 | GT1_Sucrose_synthase | 2.0e-151 | 180 | 674 | 496 | + This family is most closely related to the GT1 family of glycosyltransferases. The sucrose-phosphate synthases in this family may be unique to plants and photosynthetic bacteria. This enzyme catalyzes the synthesis of sucrose 6-phosphate from fructose 6-phosphate and uridine 5'-diphosphate-glucose, a key regulatory step of sucrose metabolism. The activity of this enzyme is regulated by phosphorylation and moderated by the concentration of various metabolites and light. | ||
| TIGR02472 | sucr_P_syn_N | 5.0e-170 | 179 | 673 | 496 | + sucrose-phosphate synthase, putative, glycosyltransferase domain. This family consists of the N-terminal regions, or in some cases the entirety, of bacterial proteins closely related to plant sucrose-phosphate synthases (SPS). The C-terminal domain (TIGR02471), found with most members of this family, resembles both bona fide plant sucrose-phosphate phosphatases (SPP) and the SPP-like domain of plant SPS. At least two members of this family lack the SPP-like domain, which may have binding or regulatory rather than enzymatic activity by analogy to plant SPS. This enzyme produces sucrose 6-phosphate and UDP from UDP-glucose and D-fructose 6-phosphate, and may be encoded near the gene for fructokinase. | ||
| TIGR02468 | sucrsPsyn_pln | 0 | 1 | 1071 | 1088 | + sucrose phosphate synthase/possible sucrose phosphate phosphatase, plant. Members of this family are sucrose-phosphate synthases of plants. This enzyme is known to exist in multigene families in several species of both monocots and dicots. The N-terminal domain is the glucosyltransferase domain. Members of this family also have a variable linker region and a C-terminal domain that resembles sucrose phosphate phosphatase (SPP) (EC 3.1.3.24) (see TIGR01485), the next and final enzyme of sucrose biosynthesis. The SPP-like domain likely serves a binding and not a catalytic function, as the reported SPP is always encoded by a distinct protein. | ||
| Gene Ontology | |
|---|---|
| GO Term | Description |
| GO:0005985 | sucrose metabolic process |
| GO:0009058 | biosynthetic process |
| Annotations - NR Download unfiltered results here | |||||||
|---|---|---|---|---|---|---|---|
| Source | Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End | Description |
| Swiss-Prot | A2WYE9 | 0 | 1 | 1078 | 1 | 1084 | SPS_ORYSI RecName: Full=Probable sucrose-phosphate synthase; AltName: Full=UDP-glucose-fructose-phosphate glucosyltransferase |
| DDBJ | BAA19241.1 | 0 | 27 | 1078 | 2 | 1047 | Sucrose-Phosphate Synthase [Saccharum officinarum] |
| GenBank | EEC72045.1 | 0 | 1 | 1078 | 1 | 1100 | hypothetical protein OsI_04951 [Oryza sativa Indica Group] |
| RefSeq | NP_001105694.1 | 0 | 1 | 1078 | 1 | 1068 | sucrose phosphate synthase1 [Zea mays] |
| RefSeq | XP_002458991.1 | 0 | 1 | 1078 | 1 | 1081 | hypothetical protein SORBIDRAFT_03g043900 [Sorghum bicolor] |
| Annotations - PDB Download unfiltered results here | |||||||
|---|---|---|---|---|---|---|---|
| Source | Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End | Description |
| PDB | 2r68_A | 0 | 202 | 673 | 31 | 453 | B Chain B, Crystal Structure Of An Interleukin-1 Receptor Complex |
| PDB | 2r66_A | 0 | 202 | 673 | 31 | 453 | B Chain B, Crystal Structure Of An Interleukin-1 Receptor Complex |
| PDB | 2r60_A | 0 | 202 | 673 | 31 | 453 | A Chain A, Structure Of Apo Sucrose Phosphate Synthase (Sps) Of Halothermothrix Orenii |
| PDB | 3s29_H | 2e-38 | 181 | 674 | 281 | 764 | A Chain A, Structure Of Apo Sucrose Phosphate Synthase (Sps) Of Halothermothrix Orenii |
| PDB | 3s29_G | 2e-38 | 181 | 674 | 281 | 764 | A Chain A, Structure Of Apo Sucrose Phosphate Synthase (Sps) Of Halothermothrix Orenii |
| Metabolic Pathways | |||
|---|---|---|---|
| Pathway Name | Reaction | EC | Protein Name |
| sucrose biosynthesis | SUCROSE-PHOSPHATE-SYNTHASE-RXN | EC-2.4.1.14 | sucrose-phosphate synthase |
| Hydropathy |
|---|
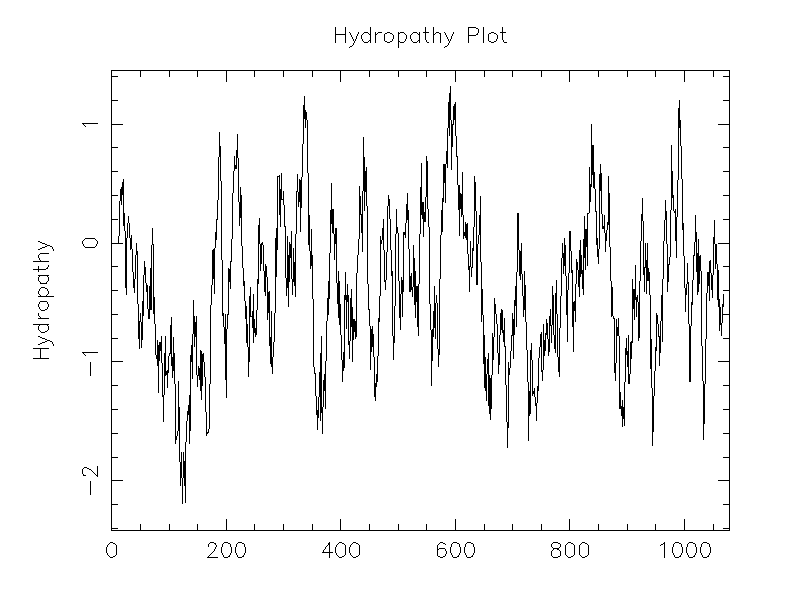 |
| EST Download unfiltered results here | ||||
|---|---|---|---|---|
| Hit | Length | Start | End | EValue |
| HO796578 | 436 | 156 | 583 | 0 |
| HO796578 | 85 | 570 | 654 | 6.99949e-42 |
| HO796578 | 27 | 663 | 689 | 6.99949e-42 |
| HO796578 | 81 | 100 | 178 | 9e-35 |
| HO796578 | 40 | 66 | 105 | 9e-35 |
| Sequence Alignments (This image is cropped. Click for full image.) |
|---|
 |
